Жалобы:
задержка психо-моторного развития, периодические рвоты.
Анамнез:
родился в срок, 3350г/ 53 см, ОША 8/9, выписан домой на 3 сутки. Голову держит с 3 мес., сидит с 6 мес., ходит с 1г (с поддержкой за руку).
1 мес.: невролог — перинатальное поражение ЦНС;
10 мес.: необъяснимые эпизоды рвот, консультирован гастроэнтерологом — нарушение моторики ЖКТ, синдром циклической рвоты?
1г 3 мес.: офтальмолог — сходящееся косоглазие;
1г 5 мес.: многократная неукротимая рвота, мама отметила, что стал хуже ходить, потерял интерес к окружающим.
Осмотр:
- реакция на осмотр негативная, светлые волосы, голубые глаза, гипертрихоз рук (со слов мамы, от отца).
- Легкое ограничение движения глазных яблок во всех направлениях, сходящееся косоглазие, спонтанный нистагм с ротаторным компонентом.
- Мышечная гипотония с элементами дистонии, рефлекс Бабинского с 2-х сторон.
- Походка неуверенная, шаткая. Фразовой речи нет.
Ранее проведенные исследования:
б/х крови: КФК 89 Ед/л, АЛТ 65 Ед/л, АСТ 44 Ед/л; лактат 3,4 ммоль/л;
УЗИ ОБП: без особенностей;
Видео-ЭЭГ мониторинг: признаки условно эпилептиформной пароксизмальной активности в задних отделах полушарий;
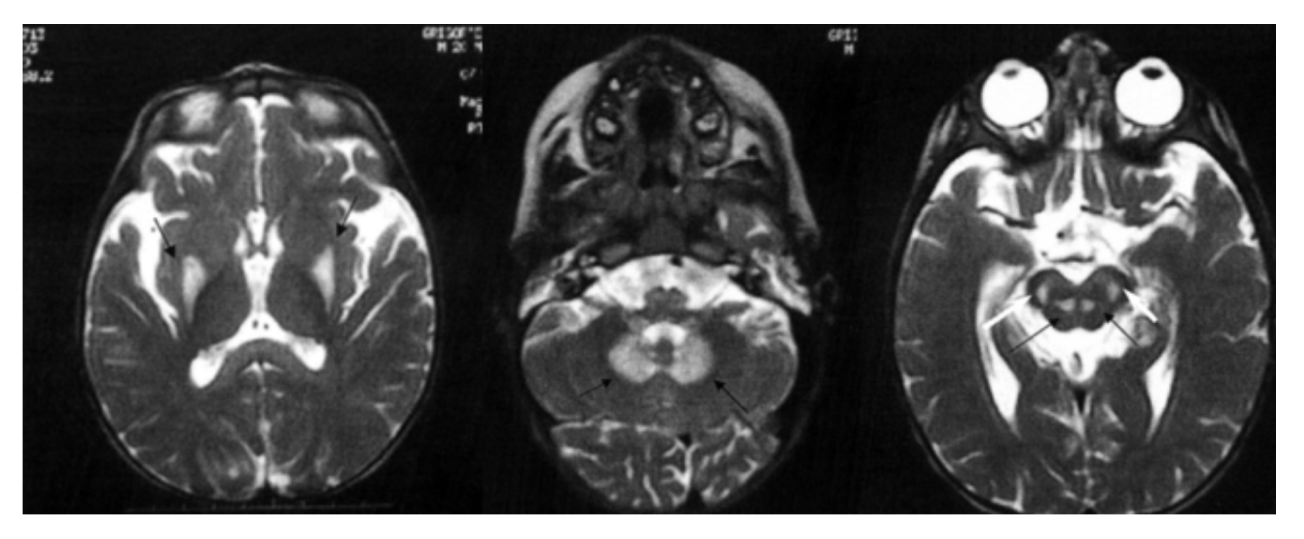
МРТ ГМ: СМОТРЕТЬ СНИМОК;
ЭНМГ: признаки смешанной полинейропатии, преобладает демиелинизирующее поражение
периферических нервов.
____________________________________________________________________________________
* Случай смоделирован. Любое совпадение с реальной клинической практикой – случайность
| Начать |
| Далее |
| Проверить |
| Показать результат |
Правильный диагноз
Синдром Ли
- В представленном случае обращают на себя внимание прогрессирующее течение заболевания в виде утраты приобретенных навыков, мышечная гипотония с элементами дистонии, мозжечковый синдром, полиневропатический синдром, гипертрихоз, повышение лактата.
- На МРТ ГМ (Т2) симметричные гиперинтенсивные очаги в области базальных ганглиев, мозжечка, красных и субталамических ядер.
Синдром Ли (подострая некротизирующая энцефаломиелопатия) — наследственное заболевание, ассоциированное с мутациями в генах, кодирующих компоненты дыхательной цепи митохондрий и белки, участвующие в их сборке.
Часто протекает под «масками» энцефаломиелита, перинатального поражения
нервной системы, нервно-мышечных заболеваний, наследственных болезней обмена.
Дебют:
- первые годы жизни (преимущественно);
- подростковый период (редко).
Основные проявления:
- прогрессирующие неврологические расстройства: утрата раннее приобретенных психомоторных навыков, мышечная гипотония, мозжечковые и экстрапирамидные расстройства, судороги;
- часто гипертрихоз (туловище, конечности);
- лактат-ацидоз;
- периферическая полинейропатия;
- эпилептический синдром;
- характерные изменения на МРТ ГМ.
б/х исследования:
- ↑ лактата в крови и спиномозговой жидкости;
- ↑ соотношения лактат/пируват;
- «парадоксальная гиперкетонемия»: ↑ уровня кетоновых тел после пищевой нагрузки и
- высокое соотношение 3-гидроксибутират/ ацетоацетат в крови;
- оргкислоты мочи: ↑ кислот, участвующих в цикле Кребса (фумаровая, янтарная и др.)
Диагностика:
- частые мутации мтДНК и ядерных генов, характерных для синдрома Ли;
- анализ органических кислот в моче;
- исследования методами NGS (панели генов, ассоциированных с наследственными митохондриальными заболеваниями; секвенирование мтДНК, экзома, генома).
Дифференциальный диагноз в группе митохондриальных энцефаломиопатий (синдром Кернс-Сейра, синдром NARP).
| Ещё раз |