
Пробанд – девочка, 9 лет
Направлена неврологом с подтвержденным диагнозом: врожденная миастения, для уточнения формы заболевания.
Жалобы: не прыгает и не бегает, трудно подниматься по лестнице, тремор рук, больше при волнении.
Анамнез: семейный не отягощен, перинатальный период без патологии, ОША 8/9, домой выписали на 3е сутки. Раннее развитие по возрасту. В 3 года обратили внимание, что не умеет прыгать и бегать. Всегда с трудом поднималась по лестнице. Получала симптоматическую терапию без выраженного положительного эффекта. В 7 лет стали замечать тремор пальцев рук.
Панельное NGS: вероятно-патогенный вариант в гене CHRNA1 (c: 190-2A>G) в гетерозиготном состоянии. Варианты в гене CHRNA1 описаны у пациентов с врожденной миастенией 1А типа.
Заключение невролога: перинатальное поражение ЦНС, синдром мышечной гипотонии. Врожденная миастения 1А в форме умеренного нижнего проксимального парапареза.
Проведенные исследования:
- антиMuSK АТ и АТ к АЦХ рецепторам в норме;
- прозериновая проба отрицательная;
- КФК 97 Ед/л
- МРТ мышц нижних конечностей: уменьшение объема мышечной ткани с обеих сторон;
- ЭНМГ: ↑ амплитуды и средней длительности ПДЕ.
Осмотр: обращают на себя внимание снижение силы проксимальных отделов конечностей до 4б, низкие коленные рефлексы, умеренно выраженные миопатические приемы при подъеме из положения сидя на полу, затрудненная походка на пятках, тремор пальцев рук.
____________________________________________________________________________________
* Случай смоделирован. Любое совпадение с реальной клинической практикой – случайность
| Начать |
О чем можно подумать?
Диагноз неверный
Диагноз неверный
Диагноз верный
| Далее |
| Проверить |
| Показать результат |
Правильный диагноз
СМА 5q
Сегодня обсудим выбор тактики ДНК-диагностики и интерпретацию полученных данных.
Дополнительные исследования после консультации:
1) Определение числа копий генов SMN1, SMN2:
0 копий экзонов 7-8 гена SMN1,
4 копии экзонов 7-8 гена SMN2 (т.е. подтверждена гомозиготная делеция экзонов 7-8 гена SMN1).
2) Анализ сегрегации варианта в гене CHRNA1: унаследован от здоровой матери (по совокупности сведений недостаточно данных о связи варианта с заболеванием)
Миастении и врожденные миастенические синдромы могут имитировать СМА 5q, однако преобладание в клинической картине проксимальной мышечной слабости и нейрогенным паттерном поражения по данным игольчатой ЭМГ (увеличение амплитуды и длительности ПДЕ), при отсутствии признаков миастении, должны насторожить в плане наличия клинического диагноза.
По результатам NGS-диагностики проводится работа по интерпретации полученных данных: изучается ген и то, какие варианты в нем приводят к заболеванию; изучается выявленный вариант, его характеристики, базы данных, научная литература. Решается вопрос о проведении дополнительных исследовании с целью подтверждения его патогенности (анализ сегрегации, функциональный анализ и др.).
Итак:
1) Самая частая причина СМА 5q – биаллельная делеция экзонов 7-8 гена SMN1 попадает под ограничения NGS-методов. Для СМА существует быстрая таргетная диагностика.
2) Определять тактику ДНК-диагностики должен врач-генетик (учитывая ограничения методов, сроки выполнения исследований, предполагаемый диагноз).
3) Интерпретировать результаты ДНК-диагностики должен врач-генетик.
Вариант, выписанный в заключение лаборатории – еще не есть подтвержденный диагноз.
В ряде случаев это может быть критично. Напомним, что для СМА 5q существует этиопатогенетическая терапия, эффективность которой тем выше, чем раньше подтвержден диагноз. Подобная ситуация может быть и для других заболеваний, поэтому крайне важно в подобных вопросах взаимодействовать с генетиками.
Дополнительные исследования после консультации:
1) Определение числа копий генов SMN1, SMN2:
0 копий экзонов 7-8 гена SMN1,
4 копии экзонов 7-8 гена SMN2 (т.е. подтверждена гомозиготная делеция экзонов 7-8 гена SMN1).
2) Анализ сегрегации варианта в гене CHRNA1: унаследован от здоровой матери (по совокупности сведений недостаточно данных о связи варианта с заболеванием)
Миастении и врожденные миастенические синдромы могут имитировать СМА 5q, однако преобладание в клинической картине проксимальной мышечной слабости и нейрогенным паттерном поражения по данным игольчатой ЭМГ (увеличение амплитуды и длительности ПДЕ), при отсутствии признаков миастении, должны насторожить в плане наличия клинического диагноза.
По результатам NGS-диагностики проводится работа по интерпретации полученных данных: изучается ген и то, какие варианты в нем приводят к заболеванию; изучается выявленный вариант, его характеристики, базы данных, научная литература. Решается вопрос о проведении дополнительных исследовании с целью подтверждения его патогенности (анализ сегрегации, функциональный анализ и др.).
Итак:
1) Самая частая причина СМА 5q – биаллельная делеция экзонов 7-8 гена SMN1 попадает под ограничения NGS-методов. Для СМА существует быстрая таргетная диагностика.
2) Определять тактику ДНК-диагностики должен врач-генетик (учитывая ограничения методов, сроки выполнения исследований, предполагаемый диагноз).
3) Интерпретировать результаты ДНК-диагностики должен врач-генетик.
Вариант, выписанный в заключение лаборатории – еще не есть подтвержденный диагноз.
В ряде случаев это может быть критично. Напомним, что для СМА 5q существует этиопатогенетическая терапия, эффективность которой тем выше, чем раньше подтвержден диагноз. Подобная ситуация может быть и для других заболеваний, поэтому крайне важно в подобных вопросах взаимодействовать с генетиками.
| Ещё раз |
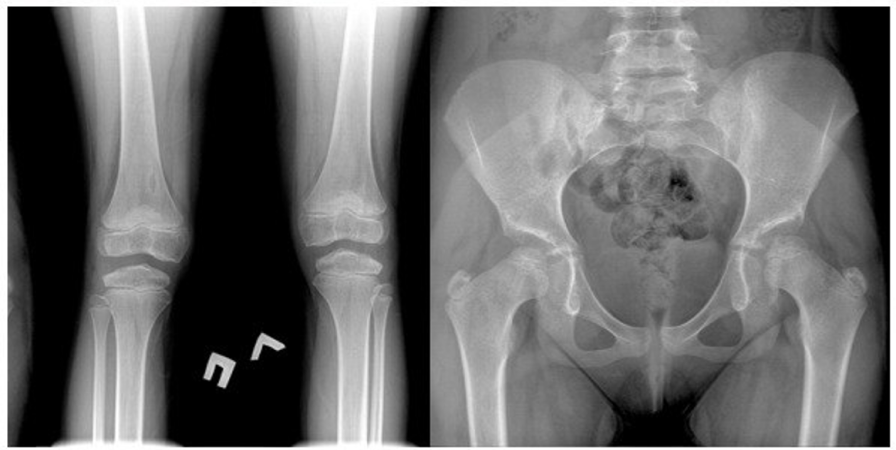
РГ тазобедренных и коленных суставов
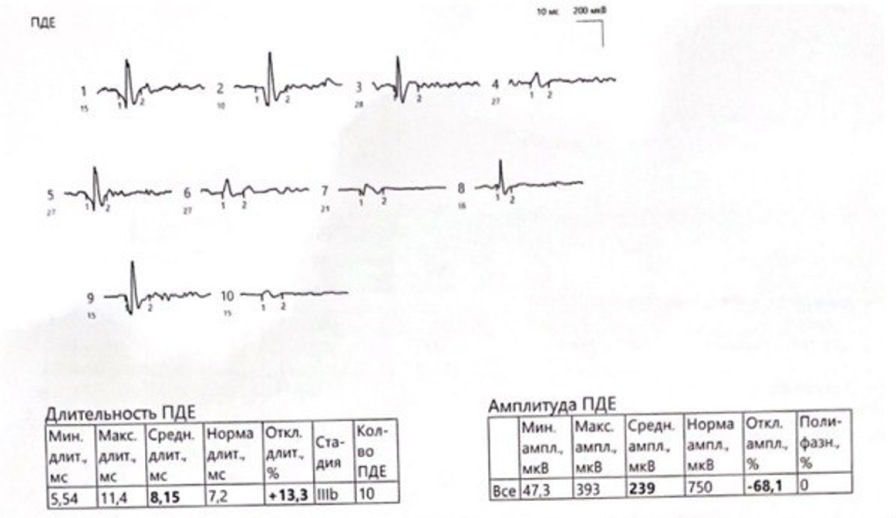
ЭМГ: выраженное уменьшение амплитуды ПДЕ
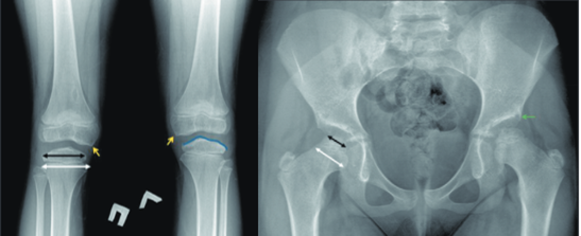
РГ-снимок коленных суставов